
Our Research
Adaptive chemistry strategies for turbulent flame simulations
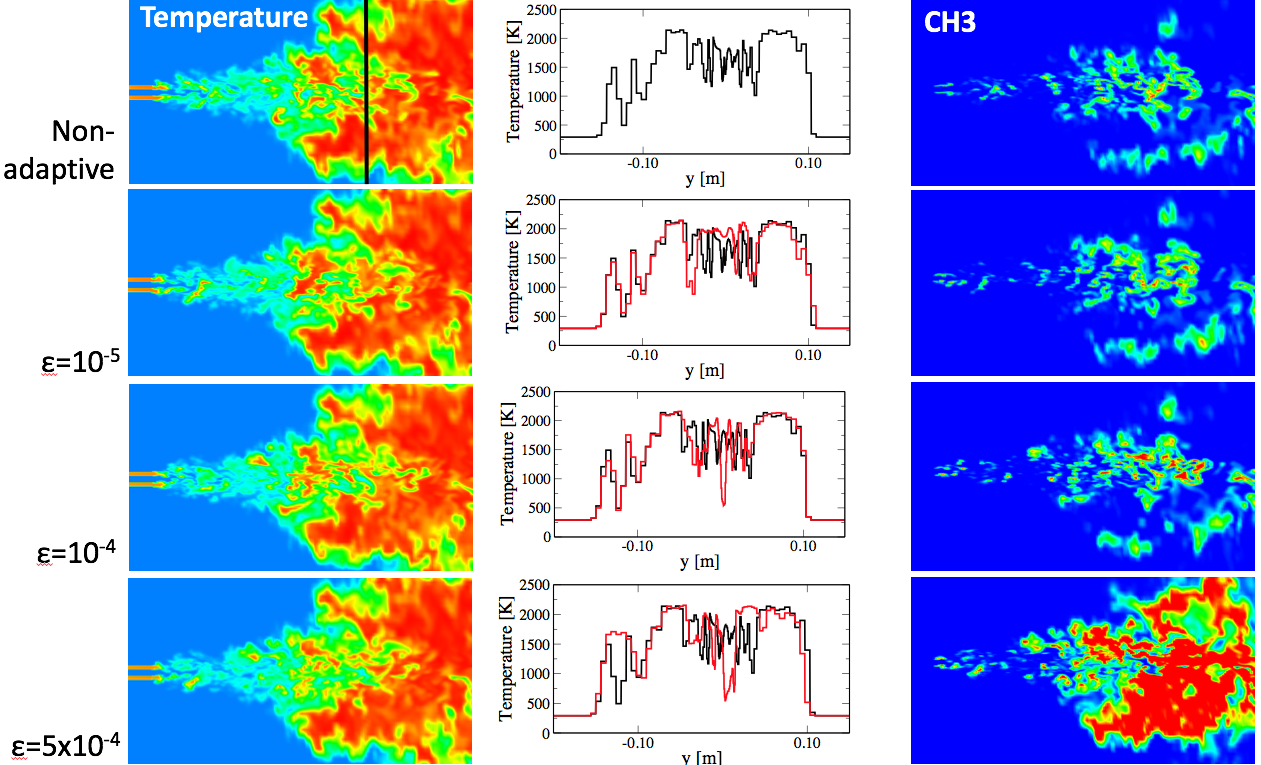
Differences in temperature and CH3 fields due to error tolerances for adaptive chemistry methods
The development of more fundamentally-based and accurate models of the chemical kinetics of real fuels has led to models involving hundreds or thousands of molecular species. A major challenge is to make use of this chemical knowledge in turbulent combustion models, which become intractable if more than tens of species are considered. However, for a given reactive flow configuration, thermodynamic conditions can vary widely in space and time, and in any small range of temperature and compositions, many species have negligible concentration, and only a few are chemically active. Hence, adapting the chemical model to local flow conditions in simulations could drastically increase the level of detail that can be afforded to describe the chemical processes.
Objective: Develop and test an adaptive chemistry strategy specifically designed for LES/PDF simulations of non-premixed turbulent flames with the following key properties: (i) locally valid reduced chemical representations are obtained through a combination of species and reaction elimination [Pepiot and Pitsch, Combust. Flame 2008], and dimension-reduction and tabulation using ISAT-RCCE [Hiremath et al., Combust. Flame 2011]; (ii) the computational particles in the LES/PDF only carry the composition variables present in the locally valid reduced representation; (iii) model reduction is performed as a pre-processing step in order to keep the run-time overhead associated with the adaptive approach to a minimum.
Numerical Investigation of Multi-component Spray Evaporation
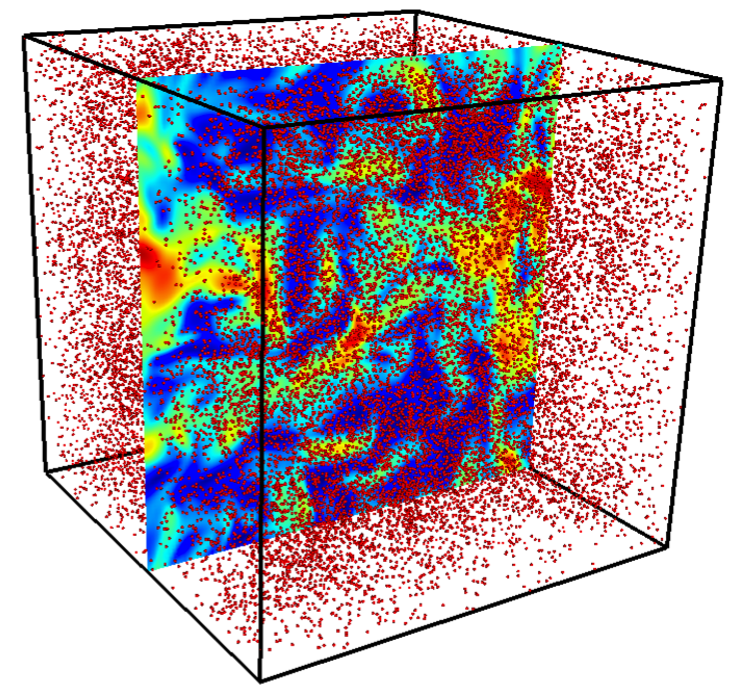
Spray droplets and scalar velocity field in a turbulent spray combustion configuration.
Turbulent spray combustion has a major role in many industrial engineering applications such as internal combustion and gas turbine engines. A good understanding of the process is vital to improving fuel efficiency, reducing emissions, and developing new alternative fuels. However, turbulent spray combustion contains both liquids and gases, making it a multiphase flow problem, and further involves a large range of coupled length and time scales and hundreds of chemical species, making it a difficult process to simulate. To obtain realistic, accurate simulations, an Euler-Lagrange strategy is used for the gas and liquid phases, with models to couple the two phases.
Objective: Investigate impact of the multi-component fuel and the evaporation model chosen on the gas phase scalar field and quantify how the evaporation model affects ignition behavior.
Thermochemical Conversion of Biomass
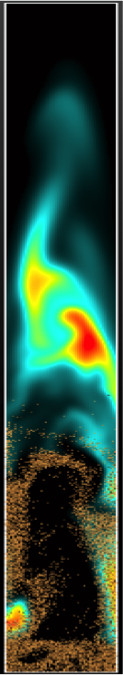
Simulation of biomass pyrolysis in a fluidized bed.
Thermochemical conversion of biomass in fluidized bed reactors (FBR) is an advanced biofuel technology that can robustly handle a wide range of feedstock and produce various types of transportation fuels. Simulations of FBR using an Euler-Lagrange approach allows the investigation of the meso-scale features of dense gas-solid flows with a relatively limited number of modeling assumptions. Of specific interest for biomass thermochemical conversion are the mixing characteristics of the flow for both phases, since they will have a first order impact on the composition of the gas exiting the reactor. However, the complexity of the multiphase, reacting, and inherently multi-scale flow severely hinders the development of reliable and accurate tools and models. Moreover, intra-particle (biomass) heat and mass transport plays a crucial role in biomass-to-biofuel conversion.
Objective:
We are using a Lagrangian numerical framework to simulate dense granular flows with compact multi-phase kinetic model (developed from detailed chemical kinetic models) to investigate the link between biomass segregation, mixing characteristics, favored chemical pathways, and hydrodynamic features of the bed. Intra-particle details of the biomass particles are also included using a 1D spherical model. Ultimately, potential alterations to the flow features will be considered to provide a better control of the chemical processes, with application to tar mitigation during gasification at high temperature, and pyrolysis oil quality control at lower temperature.
Numerical Tracers for Chemical Networks
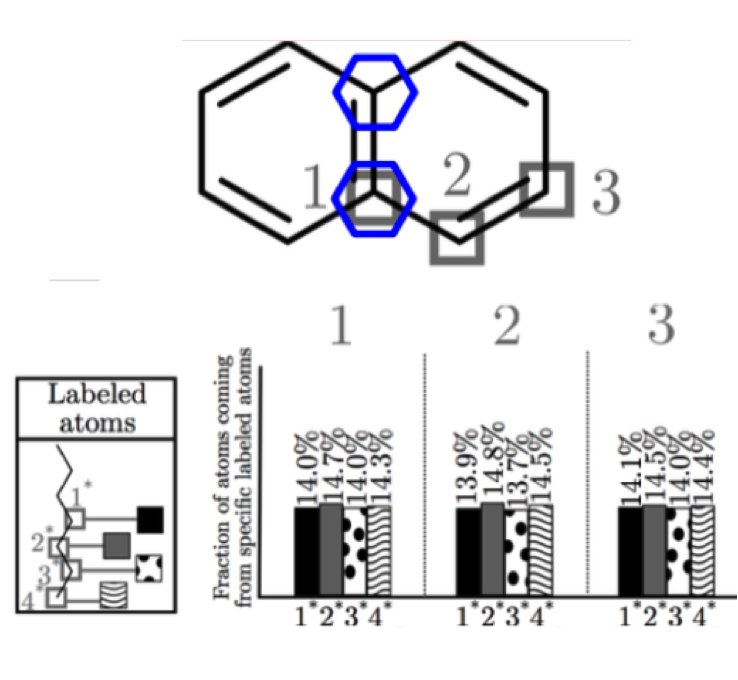
Fraction of atoms coming from specific labeled atom locations using numerical tracers method
Transportation fuels typically consist of a large number of fuel constituents, each potentially playing different roles in the combustion process. The resulting measures of combustion performance therefore result from complex multi-level physical and chemical interactions between the constituents. For instance, the sooting propensities of fuels has been shown to strongly correlate with the structural groups of the hydrocarbon fuels. Recent studies using surrogate fuels also emphasize the importance of matching the overall structural composition of a fuel.
Objective: Use a novel automated algorithm to track atoms or groups of atoms during combustion, leading to new capabilities in analyzing kinetic models for complex fuels.
Kinetic modeling of hydrocarbon fuels
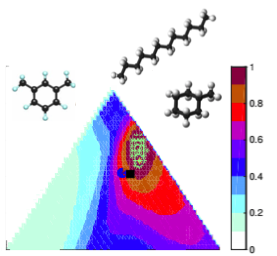
Graph of goodness of jet fuel surrogate compositions for optimization with fuel component molecules.
Using surrogate fuels in lieu of real fuels is an appealing concept for combustion studies. A major limitation however, is the capability to design compact and reliable kinetic models that capture all the specificities of the simpler, but still multi-component surrogates. This task is further complicated by the fairly large nature of the hydrocarbons commonly considered as potential surrogate components, since they typically result in extremely large detailed reaction schemes.
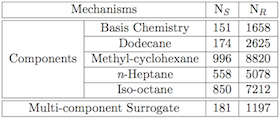
Example of jet fuel surrogate compositions and size of corresponding kinetic mechanisms.
Objective: Develop semi-automatic methods to reduced, combine, and validate kinetic models for fuel mixtures relevant to petroleum-based and biofuels. [Collaboration with K. Narayanaswamya, Stanford University, and H. Pitsch, RWTH Aachen, Germany.]

Visiting student Catherine Gruselle from CORIA (France) presents her work on stratified flame propagation at Cornell's Computational Fluid Dynamics Seminar on October 30, 2012.